The basic workflow of the rPHG2 package is as
follows:
- Create a connection object
- Read data into the R environment
- Analyze and visualize data retrieval
This document introduces you to rPHG2’s methods and
grammar, and shows you how to apply them to the previously mentioned
workflow.
Creating connection objects
rPHG objects can be created through two primary sources: * local data * server connections
Creating initial “connection” objects helps unify downstream reading and evaluation steps for PHGv2 data. In the next couple of sections, we will show you how to create either local or server connnection objects.
Local data
Local connections are for local TileDB instances or direct
locations of hVCF
files on a local disk. To create a local connection, we will create a
PHGLocalCon object using the following constructor
function:
For the above example, we will create a connection to some example
local hVCF files provided with the rPHG2 package:
LineA.h.vcfLineB.h.vcf
Since the full paths to these files will differ between each user, we
can use the system.file() function to get the full
path:
system.file("extdata", "LineA.h.vcf", package = "rPHG2")This will get the full path from the extdata directory
for the file, LineA.h.vcf, found in the rPHG2
source code.
We can further build on this to create a collection of full file paths to our hVCF data:
hVcfFiles <- system.file(
"extdata",
c("LineA.h.vcf", "LineB.h.vcf"),
package = "rPHG2"
)Now that we have a collection of hVCF files, we can use the
PHGLocalCon() constructor function to create a
PHGLocalCon object:
localCon <- hVcfFiles |> PHGLocalCon()
localCon## A PHGLocalCon connection object
## ❯ DB URI.......: ◻
## ❯ hVCF Files...: ◼Additionally, if you have a directory of valid hVCF
files, you can simply pass the directory path to the constructor. In the
following example, I will point to the extdata data
directory without pointing to singular hVCF files that is found
within the package:
system.file("extdata", package = "rPHG2") |> PHGLocalCon()## A PHGLocalCon connection object
## ❯ DB URI.......: ◻
## ❯ hVCF Files...: ◼From here, we can move to the next
section to create a HaplotypeGraph object interface
with the JVM.
Server connections
Note
We are still actively working on this section and will not have the same compatibility as rPHG. Stay tuned for further details!
Conversely, server locations are for databases served on
publicly available web services leveraging the Breeding API (BrAPI) endpoints. Since this
is a connection to a server, a URL path instead of a local file path
will be needed. We will use the PHGServerCon() constructor
function to create a PHGServerCon object:
srvCon <- "phg.maizegdb.org" |> PHGServerCon()
srvCon## A PHGServerCon connection object
## ❯ Host............: phg.maizegdb.org
## ❯ Server Status...: 200 (OK)In the above example, we have made the assumptions that this URL:
- Uses a secure transfer protocol (
https) - Uses default ports for data serving
- Has BrAPI specified endpoints
For points 1 and 2, if the URL uses
non-secure protocols (“http”) and/or has a modified port number, you
will need to specify these with the protocol and
port parameters in the constructor function. For
example:
"www.my-unsecure-phg.org" |>
PHGServerCon(
port = 5300,
protocol = "http"
)For point 3, if the constructor cannot resolve mandatory endpoints, an exception will occur.
Creating JVM objects
Note
This will require the
rJavapackage to be installed along with a modern version of Java (e.g., v17) to work properly!
Now that we have either a local or server-based connection object, we
can convert the raw hVCF data into a HaplotypeGraph
JVM object and bridge the Java reference pointer to R. Before we build
the JVM graph object, we need to initialize the JVM and add the JAR
files to our environment that are found within the latest
distribution of PHGv2.
Note: If you have not downloaded this, please see instructions here before you continue!
To initialize, run the following command:
initPhg("phg/path/to/lib")…where phg/path/to/lib is the lib directory
found within the decompressed release of PHGv2. Now that the JVM has
been initialized, we can build the JVM graph using
buildHaplotypeGraph() using the local connection object as
input:
graph <- localCon |> buildHaplotypeGraph()
graph## A HaplotypeGraph object @ 68f4865
## ❯ # of ref ranges....: 38
## ❯ # of samples.......: 2
## ❯ # of chromosomes...: 2In the above example, we took the local connection object and passed
it into a HaplotypeGraph constructor. Here, we have a basic
class that contains a pointer object where we can direct data from Java
to R:
graph |> javaRefObj()## [1] "Java-Object{net.maizegenetics.phgv2.api.HaplotypeGraph@68f4865}"Returning version and memory values
Since we need to construct an interface to a local instance of Java,
errors may arise due to several issues. Two common causes are version
issues and memory allocated to the JVM. For debugging and monitoring
purposes, we can use the jvmStats() function which creates
a instance of a JvmStats object.
javaStats <- jvmStats()
javaStats## General Stats:
## ❯ # of JARs......: 205
## ❯ Java version...: 21.0.8
## ❯ PHG version....: 2.4.70.225
##
## Memory Stats (GB):
## ❯ Max............: 0.5
## ❯ Total..........: 0.246
## ❯ Free...........: 0.202
## ❯ Allocated......: 0.044This object contains several values:
- Total number of PHGv2 JAR files added to the class path
- Your local Java version
- The current PHGv2 version added to the class path
- Current memory allocation to the JVM (recorded in gigabytes
[
GB])
Reading data
Now that we have created a HaplotypeGraph object, we can
begin reading data using the read* family
of rPHG2 functions.
Sample IDs
To return a vector of sample IDs from the graph object, we can use
the readSamples() function:
graph |> readSamples()## [1] "LineA" "LineB"Reference ranges
To return information about all reference ranges found within the
graph object, we can use the readRefRanges() function. This
will return a GRanges object which is a common data class
in the GenomicRanges
package:
graph |> readRefRanges()## GRanges object with 38 ranges and 1 metadata column:
## seqnames ranges strand | rr_id
## <Rle> <IRanges> <Rle> | <character>
## [1] 1 1-1000 * | 1:1-1000
## [2] 1 1001-5500 * | 1:1001-5500
## [3] 1 5501-6500 * | 1:5501-6500
## [4] 1 6501-11000 * | 1:6501-11000
## [5] 1 11001-12000 * | 1:11001-12000
## ... ... ... ... . ...
## [34] 2 38501-39500 * | 2:38501-39500
## [35] 2 39501-44000 * | 2:39501-44000
## [36] 2 44001-45000 * | 2:44001-45000
## [37] 2 45001-49500 * | 2:45001-49500
## [38] 2 49501-50500 * | 2:49501-50500
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengthsHaplotype IDs
To return all haplotype IDs as a “sample
reference range” matrix object, we can use the
readHapIds() function:
m <- graph |> readHapIds()
# Show only first 3 columns
m[, 1:3]## 1:1-1000 1:1001-5500
## LineA_G1 "12f0cec9102e84a161866e37072443b7" "3149b3144f93134eb29661bade697fc6"
## LineB_G1 "4fc7b8af32ddd74e07cb49d147ef1938" "8967fabf10e55d881caa6fe192e7d4ca"
## 1:5501-6500
## LineA_G1 "1b568197f6f329ec5b71f66e49a732fb"
## LineB_G1 "05efe15d97db33185b64821791b01b0f"Haplotype ID metadata
To return metadata for each haplotype ID as a tibble
object, we can use the readHapIdMetaData() function:
graph |> readHapIdMetaData()## # A tibble: 76 × 6
## hap_id sample_name description source checksum ref_range_hash
## <chr> <chr> <chr> <chr> <chr> <chr>
## 1 0eb9029f3896313aebc69… LineA haplotype … /User… Md5 39f96726321b3…
## 2 12f0cec9102e84a161866… LineA haplotype … /User… Md5 546d1839623a5…
## 3 13417ecbb38b9a159e3ca… LineA haplotype … /User… Md5 5812acb1aff74…
## 4 18498579d89483ac270e8… LineA haplotype … /User… Md5 e07d04d2fc96b…
## 5 184a72815a2ba5949635c… LineA haplotype … /User… Md5 cb86faf105b19…
## 6 1b568197f6f329ec5b71f… LineA haplotype … /User… Md5 d896e9cc56e74…
## 7 1e38bd82670c3f10982f7… LineA haplotype … /User… Md5 db22dfc14799b…
## 8 3149b3144f93134eb2966… LineA haplotype … /User… Md5 57705b1e2541c…
## 9 369464a8743d2e40ad83d… LineA haplotype … /User… Md5 66465399052d8…
## 10 3ec680649615da0685b8c… LineA haplotype … /User… Md5 347f0478b1a55…
## # ℹ 66 more rowsHaplotype ID metadata (positions)
To return positional information for each haplotype ID as another
tibble object, we can use the
readHapIdPosMetaData() function:
graph |> readHapIdPosMetaData()## # A tibble: 76 × 5
## hap_id contig_start contig_end start end
## <chr> <chr> <chr> <int> <int>
## 1 0eb9029f3896313aebc69c8489923141 2 2 49501 50300
## 2 12f0cec9102e84a161866e37072443b7 1 1 1 1000
## 3 13417ecbb38b9a159e3ca8c9dade7088 2 2 1 1000
## 4 18498579d89483ac270e8cca57f34752 1 1 16501 17500
## 5 184a72815a2ba5949635cc38769cedd0 2 2 12001 16500
## 6 1b568197f6f329ec5b71f66e49a732fb 1 1 5501 6500
## 7 1e38bd82670c3f10982f70390c599a8d 2 2 16501 17500
## 8 3149b3144f93134eb29661bade697fc6 1 1 1001 5500
## 9 369464a8743d2e40ad83d1375c196bdd 1 1 6501 11000
## 10 3ec680649615da0685b8c245e0f196e2 2 2 11001 12000
## # ℹ 66 more rowsAll hVCF data
In a majority of cases, we may need more than one piece of hVCF data.
We can read all of the prior data simultaneously as
PHGDataSet object which is an in-R-memory representation of
all hVCF data:
phgDs <- graph |> readPhgDataSet()
phgDs## A PHGDataSet object
## ❯ # of ref ranges....: 38
## ❯ # of samples.......: 2
## ---
## ❯ # of hap IDs.......: 76
## ❯ # of asm regions...: 76If this object is created, we can use the prior read*
methods to instantaneously pull out the previously mentioned R data
objects:
Filter data
In some cases, you may want to query information and focus on one
specific reference range and/or sample(s). We can
filter our PHGDataSet objects using the
filter* family of functions.
For the following examples, let’s picture our primary data as a
2-dimensional matrix of:
- samples (rows)
- reference ranges (columns)
- hap ID data (elements)
If we were to represent this as an object in R called
phgDs, it would look like the following diagram:
phgDs## A PHGDataSet object
## ❯ # of ref ranges....: 38
## ❯ # of samples.......: 2
## ---
## ❯ # of hap IDs.......: 76
## ❯ # of asm regions...: 76
…where each individual colored cell is a haplotype ID.
Filter by sample
If we want to filter based on sample ID, we can use the
filterSamples() function. Simply add the sample ID or
collection of sample IDs as a character string or a vector
of character strings, respectively. Using the prior diagram
as a reference, I will filter out anything that is not the
following:
B73Ky21Mo17
phgDs |> filterSamples(c("B73", "Ky21", "Mo17"))
Note: If samples are added to the filter collection, but are not present in the data, they will be discarded. If no samples are found, an exception will be thrown stating that no samples could be found.
Filter by reference range
If we want to filter based on sample ID, we can use the
filterRefRanges() function. Currently, this takes a
GRanges object where we can specify integer-based ranges
located in genomic regions by chromosome (i.e., seqnames).
For example, if I want to return all ranges that intersect with the
following genomic regions:
| Chromosome | Start (bp) | End (bp) |
|---|---|---|
"1" |
100 |
400 |
"2" |
400 |
900 |
I can create the following GRanges object and pass that
to the filter method:
gr <- GRanges(
seqnames = c("1", "2"),
ranges = IRanges(
c(100, 800),
c(400, 900)
)
)
phgDs |> filterRefRanges(gr)
Chaining methods
Since we have two dimensions, we can filter simultaneously by “piping” or combining filter methods in one pass:
gr <- GRanges(
seqnames = c("1", "2"),
ranges = IRanges(
c(100, 800),
c(400, 900)
)
)
phgDs |>
filterSamples(c("B73", "Ky21", "Mo17")) |>
filterRefRanges(gr)
Summarize and visualize data
In addition to parsing multiple hVCF files into R data objects via a
PHGDataSet, rPHG2 also provides functions to summarize and
visualize data.
Global values
To begin, we can return general values on the number of observations
within our dataset using the numberOf* family of
functions:
phgDs## A PHGDataSet object
## ❯ # of ref ranges....: 38
## ❯ # of samples.......: 2
## ---
## ❯ # of hap IDs.......: 76
## ❯ # of asm regions...: 76
# Get number of samples/taxa
phgDs |> numberOfSamples()## [1] 2
# Get number of chromosomes
phgDs |> numberOfChromosomes()## [1] 2
# Get number of reference ranges
phgDs |> numberOfRefRanges()## [1] 38
# Get number of haplotype IDs
phgDs |> numberOfHaplotypes()## [1] 76Get counts of unique haplotype IDs
To get the unique number of haplotypes per reference range in our
PHGDataSet object, we can use
numberOfHaplotypes() again, but set the internal parameter
byRefRange to TRUE:
phgDs |> numberOfHaplotypes(byRefRange = TRUE)## # A tibble: 38 × 7
## rr_id n_haplo seqnames start end width strand
## <chr> <int> <fct> <int> <int> <int> <fct>
## 1 1:1-1000 2 1 1 1000 1000 *
## 2 1:1001-5500 2 1 1001 5500 4500 *
## 3 1:11001-12000 2 1 11001 12000 1000 *
## 4 1:12001-16500 2 1 12001 16500 4500 *
## 5 1:16501-17500 2 1 16501 17500 1000 *
## 6 1:17501-22000 2 1 17501 22000 4500 *
## 7 1:22001-23000 2 1 22001 23000 1000 *
## 8 1:23001-27500 2 1 23001 27500 4500 *
## 9 1:27501-28500 2 1 27501 28500 1000 *
## 10 1:28501-33000 2 1 28501 33000 4500 *
## # ℹ 28 more rowsVisualize counts of unique haplotype IDs
We can visualize the prior section using a member of the
plot* family of functions, plotHaploCounts().
By default, this will produce a scatter plot of data found across all
reference ranges in the reference genome (similar to a Manhattan
plot):
phgDs |> plotHaploCounts()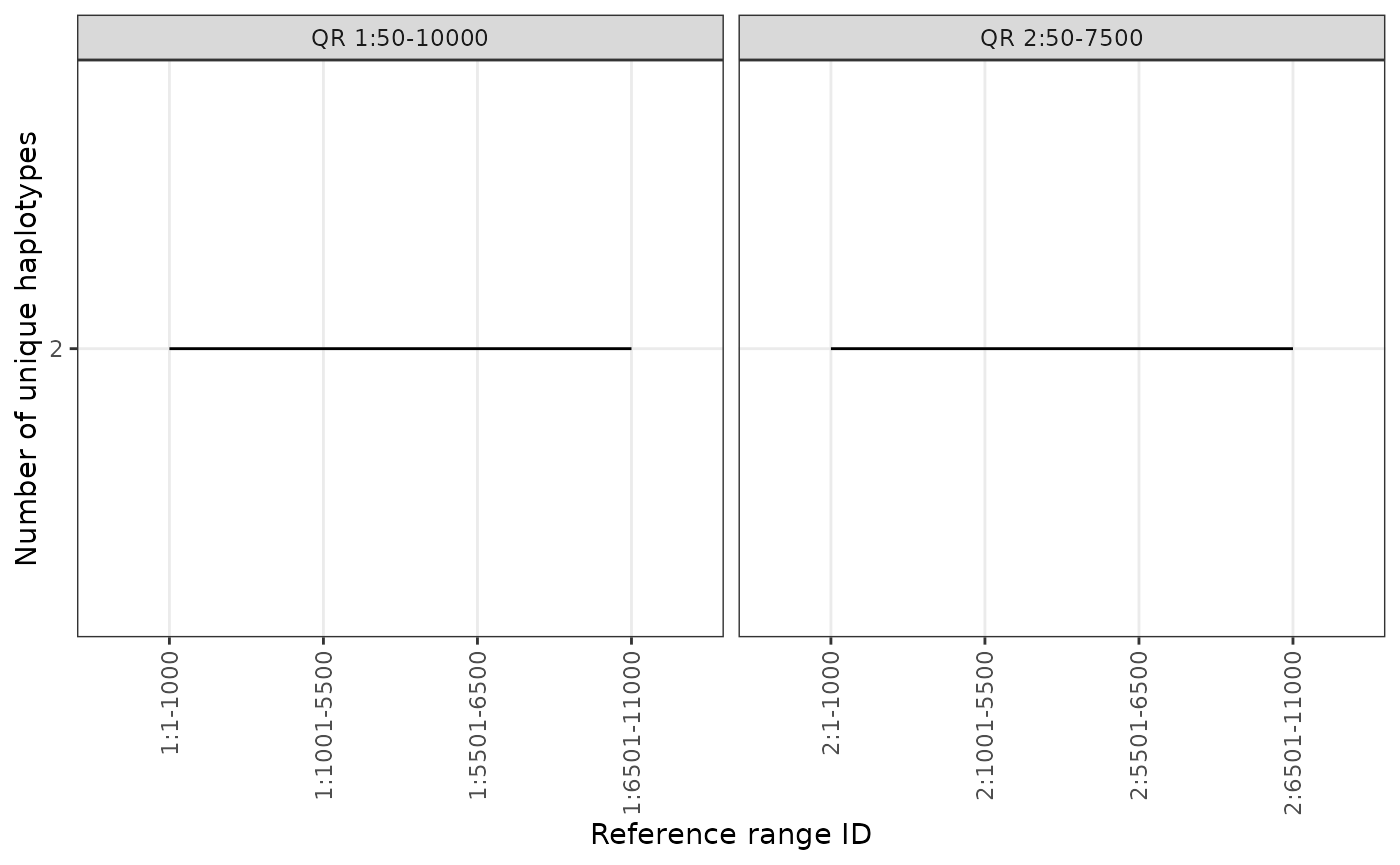
To get more “granular” views of specific regions within our data, we
can pass a GRanges object from the package GenomicRanges
to the gr parameter:
# library(GenomicRanges)
query <- GRanges(
seqnames = "1",
ranges = IRanges(start = 50, end = 10000)
)
phgDs |> plotHaploCounts(gr = query)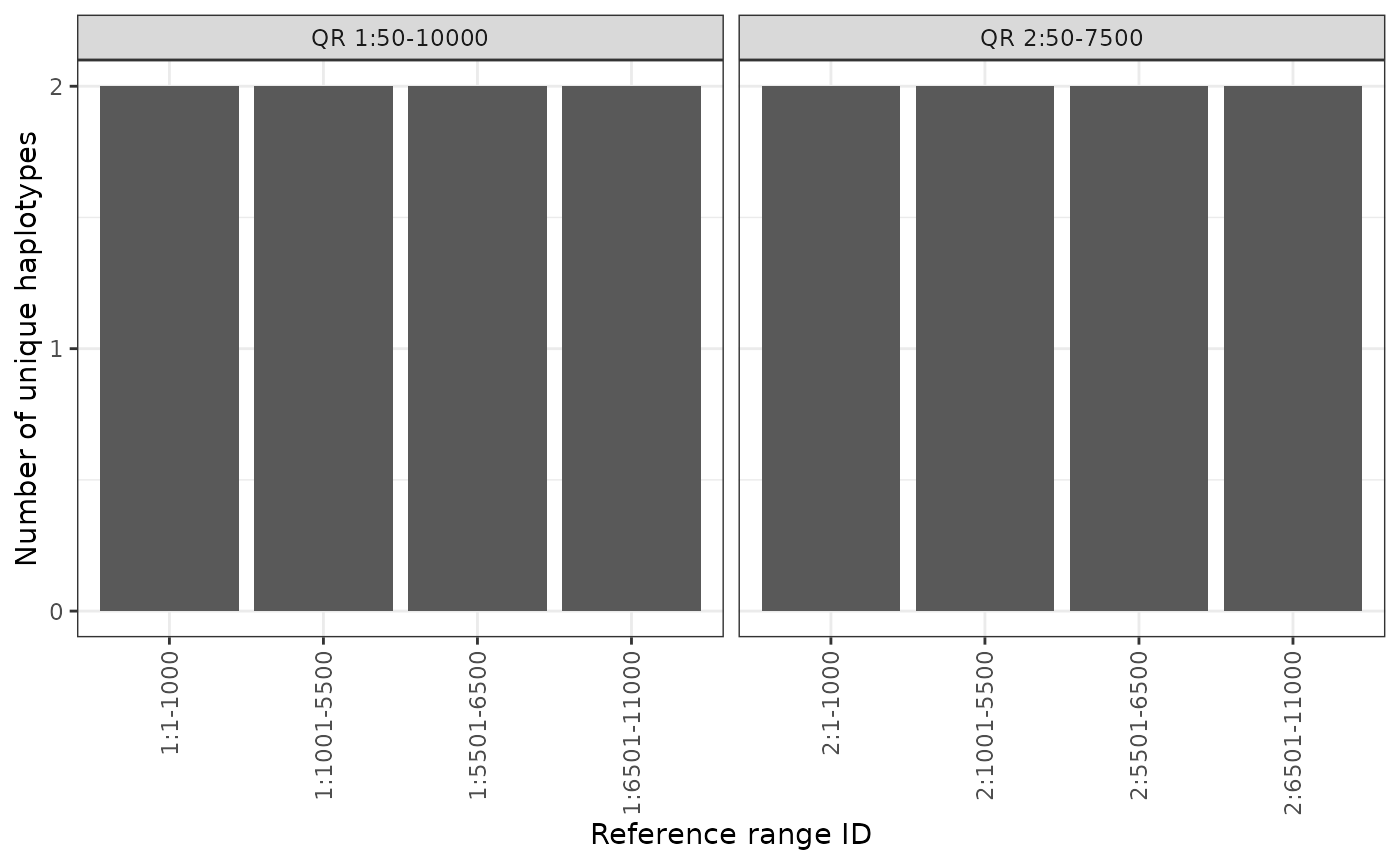
We can also query mutltiple regions of interest simultaneously by
adding more observations to the query object:
# library(GenomicRanges)
query <- GRanges(
seqnames = c("1", "2"),
ranges = IRanges(start = c(50, 50), end = c(10000, 7500))
)
phgDs |> plotHaploCounts(gr = query)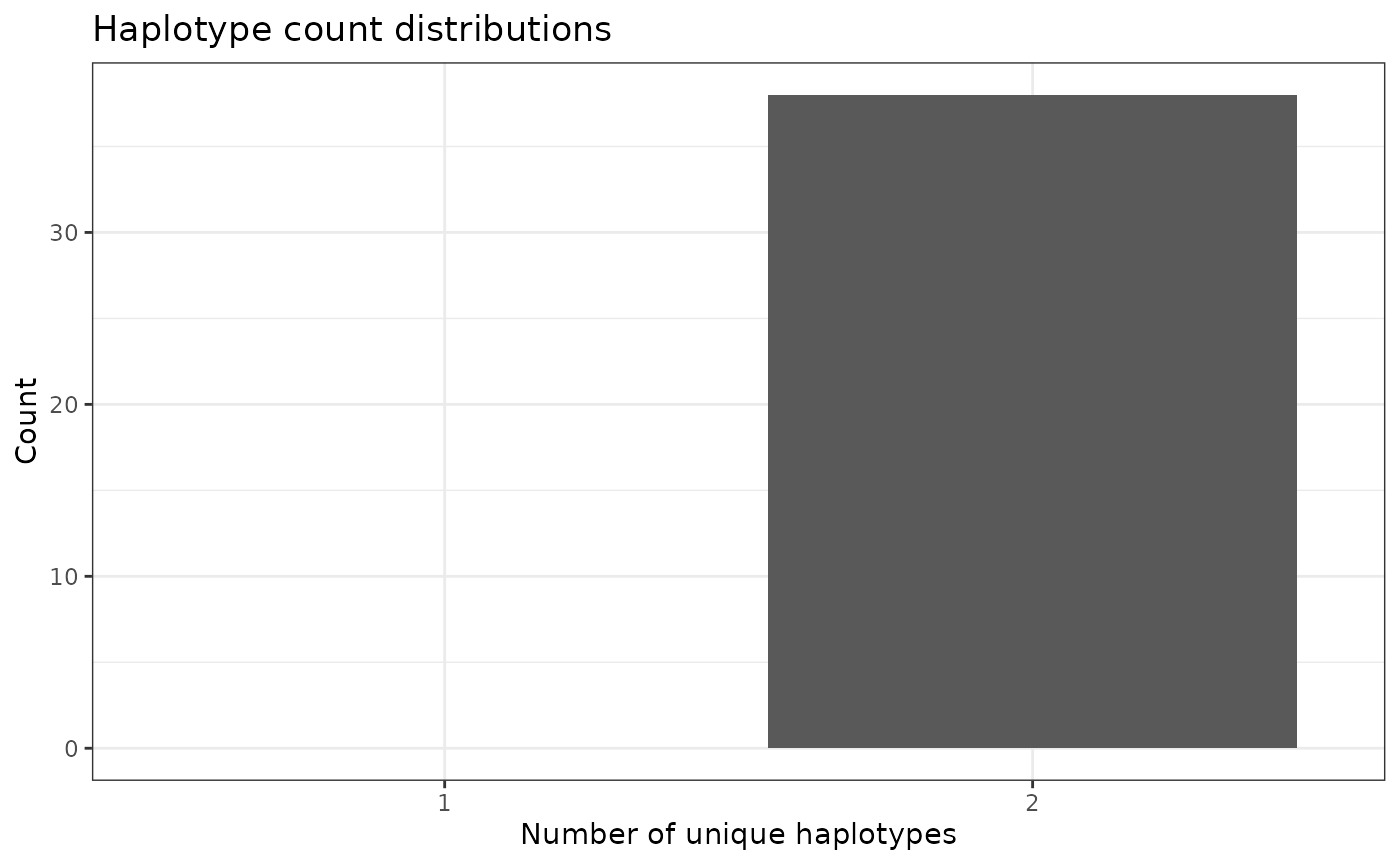
Finally, we provide 3 separate “geometry” options for plotting using
the geom parameter: * "l" - line plotting
(e.g., geom_line()) (default) * "b" -
bar plotting (e.g., geom_bar()) * "p" - point
plotting (e.g., geom_point())
For example, if we want to switch this from a line plot to a bar
plot, we can specify geom = "b":
phgDs |> plotHaploCounts(gr = query, geom = "b")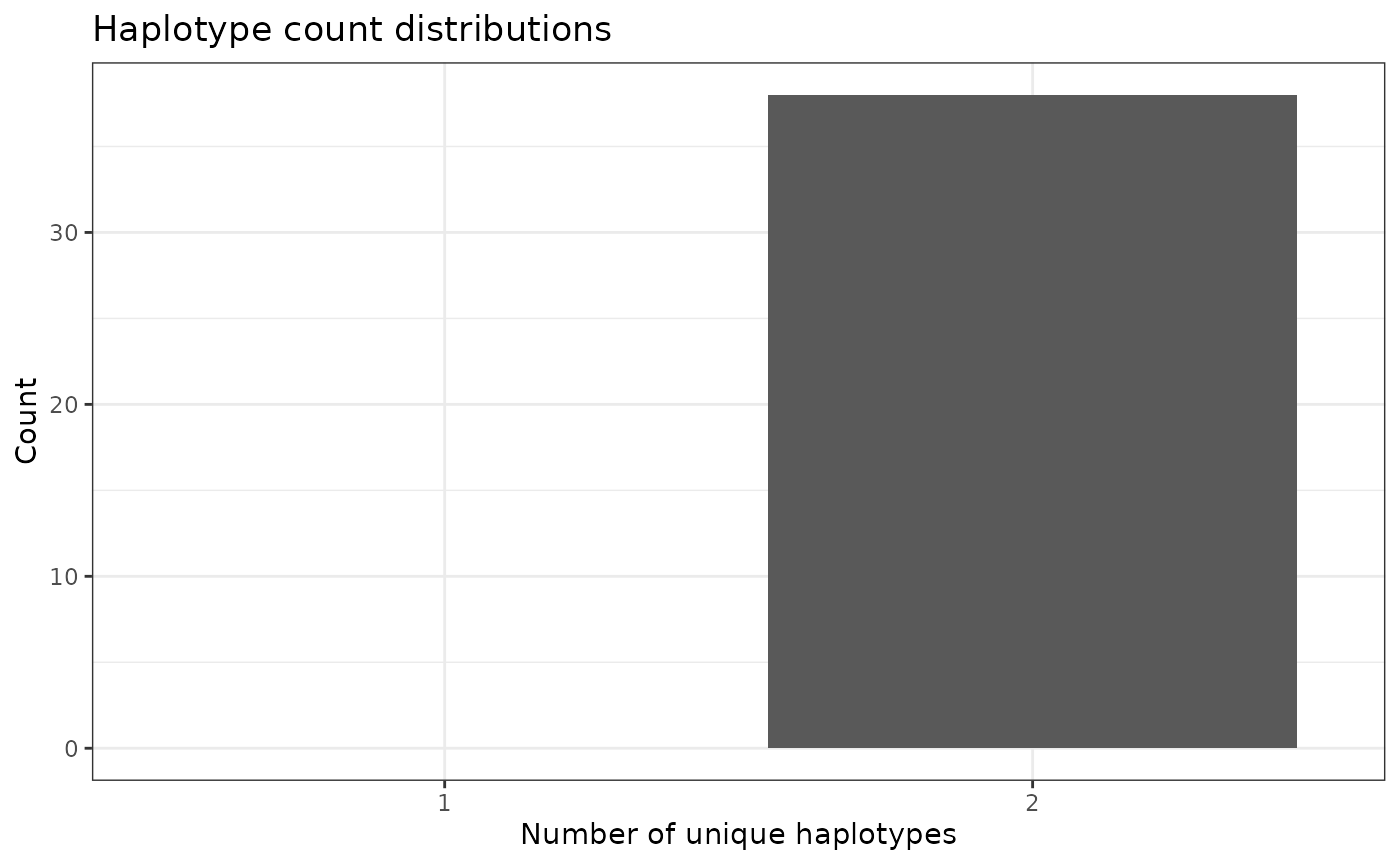
Visualize distributions of unique haplotype IDs
To get a more “global” estimate of unique haplotype ID distribution
across the genome, we can us the function
plotHaploDist():
phgDs |> plotHaploDist()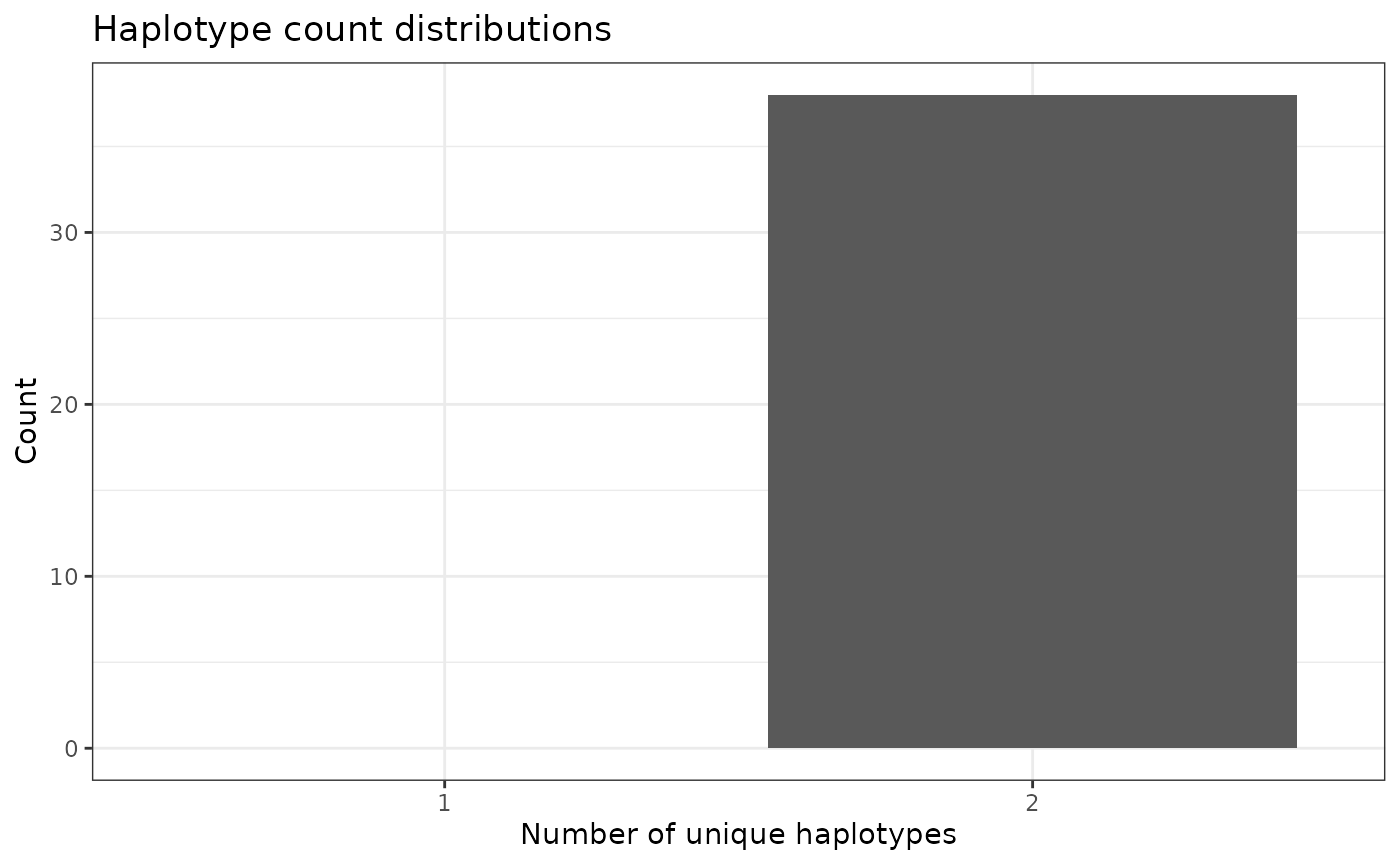
Returning sequence data
We can also return haplotype sequence information via the
readSequence() command with PHG data sets (i.e.,
PHGDataSet objects) pointed to PHGv2
databases containing compressed assemblies information.
In order to perform sequence retrieval, we must perform a couple of steps:
- Add
hostinformation to aPHGLocalConobject - Add an AGC path to the global options (i.e., the
options()function)
Setup - add host information
For step 1, during the the construction of a
PHGLocalCon object using the PHGLocalCon()
function, add the path to a PHGv2
database containing a compressed sequence file:
dbPath <- "/my/phgv2/db_path/"
# using 'hVcfFiles' variable from earlier steps
localCon <- hVcfFiles |> PHGLocalCon(dbUri = dbPath)If you are unsure what this path is, it should be the directory that was created during the PHGv2 building and loading steps and contains files and sub-directories that looks like the following:
phg_v2_example/
├── data
│ ├── anchors.gff
│ ├── annotation_keyfile.txt
│ ├── Ref-v5.fa
│ ├── LineA-final-01.fa
│ └── LineB-final-04.fa
├── output
│ └── updated_assemblies
│ ├── Ref.fa
│ ├── LineA.fa
│ └── LineB.fa
└── vcf_dbs
├── assemblies.agc *
├── gvcf_dataset/
├── hvcf_dataset/
└── temp/Note
In order for this to work, this directory/database must contain a compressed sequence file. By default, this file will be called
assemblies.agc. This is a compressed file generated by the Assembled Genomes Compressor.
Setup - add AGC path
For step 2, we must also have a working AGC application for our operating system and environment. This can be established in a couple of ways:
Option 1 - Conda
If you went through the steps of setting up a PHG database on your working machine, we can assume that you have:
- A working Conda installation
- A working PHGv2 Conda environment. This by default
will be called
phgv2-conda.
If this is the case, you will, by default, have AGC already set up on your machine since this is a necessary application for several crucial PHGv2 steps. You can verify AGC is present in your PHGv2 environment by running the following command within a terminal:
conda run -n phgv2-conda agcIf present, this will output a series of commands used by the
agc program. For example:
$ conda run -n phgv2-conda agc
AGC (Assembled Genomes Compressor) v. 3.1.0 [build 20240312.1]
Usage: agc <command> [options]
Command:
create - create archive from FASTA files
append - add FASTA files to existing archive
getcol - extract all samples from archive
getset - extract sample from archive
getctg - extract contig from archive
listref - list reference sample name in archive
listset - list sample names in archive
listctg - list sample and contig names in archive
info - show some statistics of the compressed data
Note: run agc <command> to see command-specific optionsNote
If you have changed the name of your environment, the value given with the
-nflag will have to change!
If your machine matches the prior criteria, you may use the rPHG2
function linkCondaBin() to link your Conda
environment’s AGC application path to R’s global options:
linkCondaBin(condaPath = "/path/to/my/conda/installation")This function will assume your environment name is called
phgv2-conda. If you have a different, you can add the
modified id to the envName parameter.
linkCondaBin(
condaPath = "/path/to/my/conda/installation",
envName = "phgv2-sorghum" # example Conda environment name
)Option 2 - manual AGC path
If you do not have Conda installed or no PHGv2 instance,
but have AGC installed on your
machine, you can manually set the path using R’s
option() function. In order for rPHG2 to correctly identify
the correct path, you must make a new key in the global options called
phgv2_agc_path:
options("phgv2_agc_path" = "/path/to/my/agc/application/bin/agc")Note
This must be the path to the AGC binary (i.e., the file named
agc) found in the release.
Setup - build PHGDataset object
Now that all prerequisites are completed, we can finish building the
PHGDataSet object using conventional rPHG2 syntax:
phgDs <- localCon
buildHaplotypeGraph() |>
readPhgDataSet()
phgDs## A PHGDataSet object
## ❯ # of ref ranges....: 38
## ❯ # of samples.......: 2
## ---
## ❯ # of hap IDs.......: 76
## ❯ # of asm regions...: 76Using readSequence()
Now that we have our PHGDataSet object created, we can
return specific sequence information relating to an individual haplotype
or a collection of haplotypes using a haplotype or reference range
identifier, respectively. Similar to other syntax, we can simply pipe
our PHGDataSet object into the readSequence()
method.
For starters, I will return a single haplotype sequence using the
hapId parameter:
# Return first haplotype ID in matrix (example)
hapId <- phgDs |> readHapIds() |> _[1, 1]
phgDs |> readSequence(hapId = hapId)## DNAStringSet object of length 1:
## width seq names
## [1] 1000 GCGCGGGGACCGAGAAACCCGGC...CCACGCACAGGCGAGGGAACGGC 1 sampleName=Line...The data object that is returned from readsequence() is
a dnastringset object which is from the Biostrings
package found on bioconductor. you can think of this object a list
of dnastringset objects which are memory-efficient
representations of biological sequences (e.g., dna). this will allow
users the ability to access all Biostrings-related methods
for objects of type dnastring. For more information about
how to use these objects, take a look at the “Quick
Overview” documentation found in the Biostrings
package.
If we are interested in returning haplotype sequence data from an
entire reference range, we can specify a reference range ID using the
rrId parameter. This identifier will be a
character object containing the following pattern:
<chromosome_id>:<start_bp>-<end_bp>For example, this identifier will look like the following “first” reference range found in our example hVCF data:
# Return first reference range in matrix (example)
rrId <- phgDs |> readHapIds() |> colnames() |> _[1]
rrId## [1] "1:1-1000"
phgDs |> readSequence(rrId = rrId)## DNAStringSet object of length 2:
## width seq names
## [1] 1000 GCGCGGGGACCGAGAAACCCGGC...CCACGCACAGGCGAGGGAACGGC 1 sampleName=Line...
## [2] 1000 GCGCGGGGACCGTGAAACCCGGC...CCACGCACAGGCGAGGTAACGGC 1 sampleName=Line...